A new European research initiative is bringing together experts, patients, and clinical centers to expand knowledge about ADTKD—the third most common genetic kidney disease. Prof. Dr. Jan Halbritter of Freiburg presented the ADTKD-Net consortium at this year’s European Renal Association (ERA) Congress in Glasgow. This European collaborative platform collects long-term data and biological samples from affected patients. The goal is to better understand the disease’s progression and to prepare for future clinical trials.
ADTKD has a 50% chance of being inherited by the next generation. The most common causes are pathogenic mutations in the UMOD and MUC1 genes, which account for the majority of cases.
COMMON, BUT ALSO OFTEN OVERLOOKED
Although ADTKD is considered the third most common hereditary kidney disease after autosomal dominant polycystic kidney disease (ADPKD) and Alport syndrome, the condition remains largely unknown among both healthcare professionals and patients. According to Prof. Halbritter, one of the reasons is the lack of clear clinical signs. “ADTKD is clinically very nonspecific,” the expert explained. The symptoms are often uncharacteristic and usually develop slowly over the course of years. Progressive deterioration of kidney function can occur without any clear warning signs. Unlike other chronic kidney diseases (CKD), large kidney cysts or severe proteinuria—which would be noticeable during routine examinations—do not typically occur. Therefore, genetic testing is essential for an accurate diagnosis.
NETWORK AIMS TO HELP CLARIFY UNANSWERED QUESTIONS
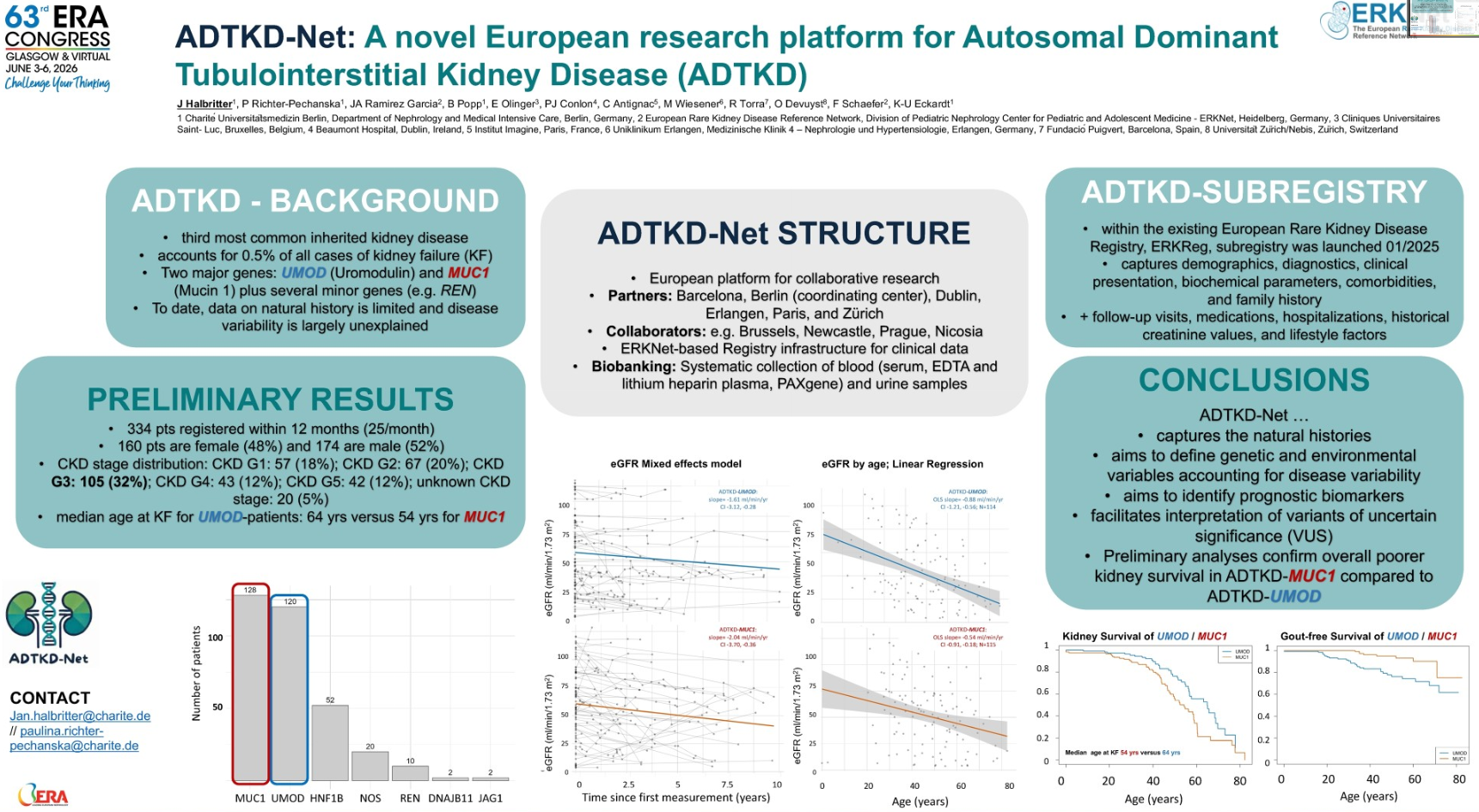
“Currently, there is very little data on the natural course of the disease. Closing this knowledge gap is the consortium’s main goal,” said Prof. Halbritter. ADTKD-Net brings together numerous academic centers across Europe. The initiative is coordinated by Charité – Universitätsmedizin Berlin, with institutions in Spain, Ireland, France, and Switzerland, as well as many other collaborating partners.
The ADTKD Registry is integrated into the infrastructure of the European Reference Network for Rare Kidney Diseases (ERKNet). It contains both prospective follow-up data and retrospective information. Not only clinical information is collected, but also blood serum, DNA, RNA, and urine samples. The biobank is primarily intended to support biomarker research.
ADTKD-Net aims to help answer important unresolved questions:
- Why does kidney failure progress rapidly in some ADTKD patients, while it remains stable for decades in others?
- Which prognostic biomarkers are associated with rapid progression?
- Which patients could benefit most from future therapies?
MORE THAN 300 PATIENTS ALREADY ENROLLED
Although the project was launched only recently, valuable data is already available. “In about a year and a half, we have enrolled 330 ADTKD patients with clinical data sets,” reported Prof. Halbritter.
The majority of registered participants have MUC1- or UMOD-associated ADTKD. The gender ratio between men and women is nearly equal. Most patients are currently in stage 3 CKD. This is relevant because the researchers are primarily interested in identifying patients before they reach kidney failure.
IMPORTANT DIFFERENCES BETWEEN GENETIC SUBTYPES
One of the first analyses focused on the decline in kidney function, as determined by the decrease in estimated glomerular filtration rate (eGFR). The results confirm that not all forms of ADTKD behave the same way. In MUC1-associated disease, kidney function appears to decline more rapidly on average than in ADTKD-UMOD. This difference was also evident in the time to kidney failure. Accordingly, the median age at which patients with ADTKD-MUC1 develop kidney failure is approximately 54 years, whereas in ADTKD-UMOD, this occurs about ten years later. “These results confirm independent data previously published from U.S. registries and, in some cases, from European patients,” summarized Prof. Halbritter.
GOUT AS A WARNING SIGN FOR THE ADTKD-UMOD SUBTYPE
Furthermore, another well-known observation was confirmed: Early-onset gout is significantly more common in UMOD-associated ADTKD than in MUC1. Prof. Halbritter highlighted the clinical implications: “This is a key indicator that should alert you to ADTKD, especially if it runs in the family,” he said in his appeal to treating physicians. If multiple family members have gout in conjunction with impaired kidney function, genetic testing should be considered. This could shorten a long diagnostic journey for affected patients, according to Prof. Halbritter.